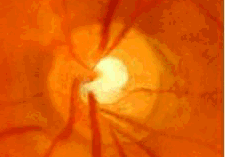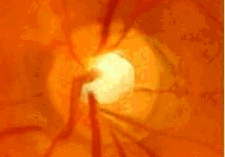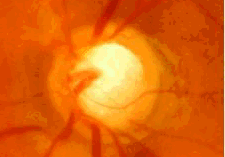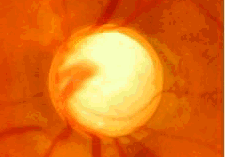
- Por defectos del desarrollo del iris: síndrome de Axenfeld- Rieger, anomalía de Peters, aniridia.
- Atrofia iridiana con alteraciones corneales: síndrome ICE, distrofia posterior polimorfa
- Glaucoma pigmentario
- Iridosquisis
- Síndrome exfoliativo
- Glaucoma neovascular
- Tumores del iris
- Uveítis anterior
- Traumas
- Complicaciones de cirugía ocular
Síndrome de Axenfeld-Rieger
 Es una rara disgenesia iridocorneal ocular congénita que involucra las estructuras angulares. Dichos cambios morfológicos conllevan a la presentación del glaucoma. Frecuentemente las alteraciones oculares coexisten con otros hallazgos sistémicos que permiten caracterizar el síndrome
Es una rara disgenesia iridocorneal ocular congénita que involucra las estructuras angulares. Dichos cambios morfológicos conllevan a la presentación del glaucoma. Frecuentemente las alteraciones oculares coexisten con otros hallazgos sistémicos que permiten caracterizar el síndrome
Características
Anomalías oculares:
- Embriotoxon posterior
- Atrofia de iris
- Corectopia
- Policoria
Anomalías no oculares: que comprometen principalmente el tercio medio facial
Anomalía de Peters
 Es un síndrome que se ha relacionado, en la mayoría de los casos, a una mutación genética. Algunos estudios demuestran su asociación a otros trastornos genéticos. La mayoría de los casos son esporádicos, pero también se han descrito patrones de herencia autosómica dominante y recesiva.
Es un síndrome que se ha relacionado, en la mayoría de los casos, a una mutación genética. Algunos estudios demuestran su asociación a otros trastornos genéticos. La mayoría de los casos son esporádicos, pero también se han descrito patrones de herencia autosómica dominante y recesiva.
En el 80% de los casos se presenta de forma bilateral, aunque asimétrica
A nivel corneal se observa opacidad del estroma posterior, Descemet y endotelio, con la periferia corneal relativamente transparente. El iris presenta adherencias a la periferia del leucoma
Pueden aparecer otras alteraciones oculares, como:
- Sinequias posteriores
- Microcórnea
- Microftalmía
- Córnea plana
- Esclerocórnea
- Coloboma de iris
- Cristalino adherido a endotelio corneal
- Persistencia de vítreo primario hiperplásico
- Glaucoma (50% al 70% de los pacientes)
Pueden aparecer alteraciones sistémicas como:
- Hidrocefalia
- Hipoplasia pulmonar
- Fisura palatina
- Malformaciones cardiacas
- Malformaciones genitourinarias
- Disóstosis craneofacial
- Alteraciones neurológicas
Aniridia
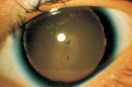 La aniridia es la ausencia clínica del iris (aunque es visible una traza circular de tejido mediante gonioscopía). Es una enfermedad congénita y hereditaria. Se trasmite al 50% de los descendientes de una forma autosómica dominante
La aniridia es la ausencia clínica del iris (aunque es visible una traza circular de tejido mediante gonioscopía). Es una enfermedad congénita y hereditaria. Se trasmite al 50% de los descendientes de una forma autosómica dominante
Manifestaciones sistémicas
Puede cursar con otras alteraciones sistémicas más excepcionales como afectaciones renales o retraso mental.
En el 20% de los casos se asocia a nefroblastoma (tumor de Wilms), especialmente cuando es bilateral. Se observan otras alteraciones congénitas como malformaciones genitourinarias y retraso mental en el 15% de los casos, causando el Síndrome de WAGR (Wilms, aniridia, genitourinario, retraso)
Síndrome de dispersión pigmentaria
Cuadro infrecuente que se caracteriza por el depósito de gránulos de pigmento sobre el SA, sobre todo en el endotelio corneal formando el huso de Krukenberg, además se deposita en el ecuador del cristalino lo cual es visible en biomicroscopía con midriasis.
Se libera más pigmento que lo normal, pero no hay glaucoma. Puede verse como secuela de traumas o cirugías. El 10% de los pacientes con SDP desarrollan glaucoma crónico de ángulo abierto
Puede verse en:
- Glaucoma pigmentario
- Miopias
- Pseudoexfoliación del cristalino
- Uveitis de larga evolución
- Puede observarse una forma secundaria de SDP y glaucoma pigmentario en ojos seudofáquicos en los que las asas del LIO rozan la cara posterior del iris
Características generales
- Hombre (lo más frecuente)
- Joven (20-40 años)
- Miopía de grado variable
- Blanco
Glaucoma pigmentario
Características oculares
- A la gonioscopía es un ángulo abierto
- Depósito de pigmento en el endotelio corneal en forma de huso vertical (huso de Krukenberg)
- CA frecuentemente profunda, sobre todo en la periferia media, en donde el iris tiende a incurvarse hacia atrás.
- La superficie anterior del iris está recubierta por finos gránulos de pigmento sobre todo en los surcos iridianos.
- Desaparición del epitelio pigmentario de la periferia del iris que da lugar a defectos en hendidura visibles por
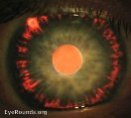 transiluminación.
transiluminación. - Se puede observar heterocromía leve.
- Depósitos de pigmento sobre ambas caras del cristalino y la zónula. En la superficie posterior del cristalino.
- En ocasiones hay depósitos de pigmento en la extrema periferia de la retina
Características gonioscópicas
- Ángulo abierto amplio

- Concavidad posterior del iris
- Acumulo de pigmento en la malla trabecular
- Pigmentación de la línea de Schwalbe de forma sinuosa (línea de Sampaolesi)
- Banda ancha del cuerpo ciliar
- Presencia de abundantes procesos iridianos

Patogenia
La dispersión del pigmento se debe al roce mecánico entre la capa posterior pigmentada del iris y la superficie anterior de las fibrillas zonulares como consecuencia de la excesiva incurvación del iris periférico en sentido posterior.
La elevación de la PIO parece deberse a obstrucción y lesión trabecular por parte del pigmento
Diagnóstico diferencial con:
GPAA con ángulos hiperpigmentados (no huso de Krukenberg, ni defectos iridianos por transiluminación)
Glaucoma por seudoexfoliación (pacientes entre 60 y 70 años en el momento del diagnóstico, algunos pueden presentar huso de Krukenberg pero los defectos iridianos por transiluminación son más manifiestos en el borde pupilar que en la periferia)
Tratamiento
Médico
Igual que en el GPAA, sólo evitar mióticos en pacientes jóvenes ya que pueden elevar la miopía al inducir espasmo ciliar
Quirúrgico
- Trabeculoplastia con láser argón (TLA)
- Trabeculoplastia Selectiva (SLT)
- Cirugía filtrante, necesaria en pacientes que no reaccionan a los tratamientos anteriores
Iridosquisis
Características
- Separación de capas iridianas
- Predomina en nasal-inferior
- En mujeres entre 60-70 años
- Atrofia iridiana
- 50% de los casos, desarrollan glaucoma por cierre angular