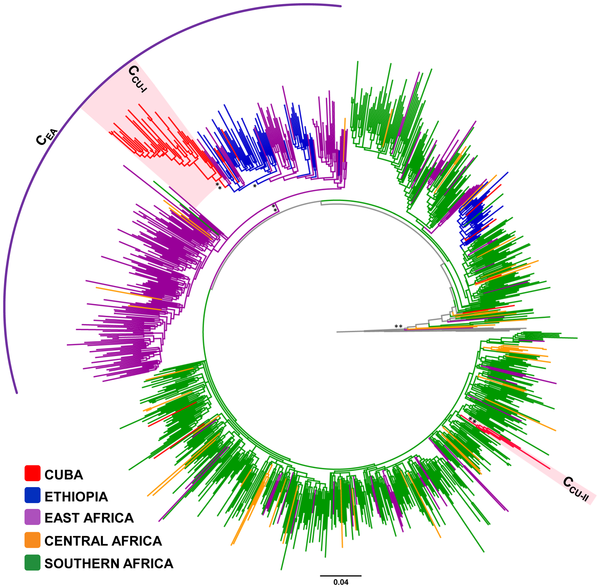
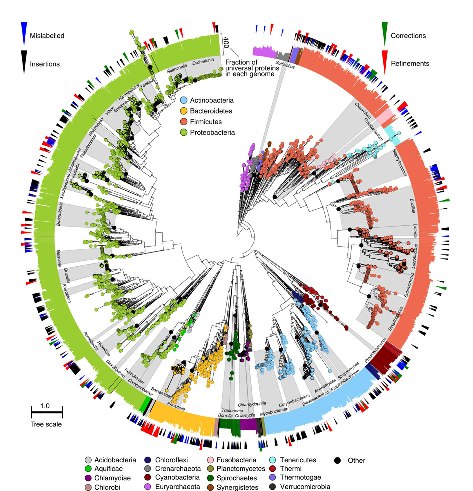
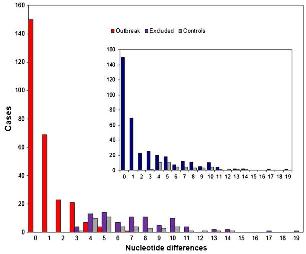
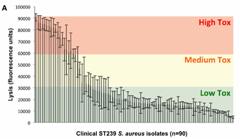
 Los datos de secuencia pueden ser parámetros clínicos relevantes para predecir la virulencia de cepas bacterianas circulantes. Tal es la conclusión del estudio del genoma de 90 cepas de estafiloco dorado resistente a la meticilina, publicado en Laabei M, Recker M, RudkiN JK, Aldeljawi M, Gulay Z, Sloan TJ, et al. Predicting the virulence of MRSA from its genome sequence. Genome Res. April 9 2014; doi: 10.1101/gr.165415.113.
Los datos de secuencia pueden ser parámetros clínicos relevantes para predecir la virulencia de cepas bacterianas circulantes. Tal es la conclusión del estudio del genoma de 90 cepas de estafiloco dorado resistente a la meticilina, publicado en Laabei M, Recker M, RudkiN JK, Aldeljawi M, Gulay Z, Sloan TJ, et al. Predicting the virulence of MRSA from its genome sequence. Genome Res. April 9 2014; doi: 10.1101/gr.165415.113.
Filed under Diagnóstico, Filogenética, Otros Proyectos Genoma, Patógenos by on . Comment. ![]()