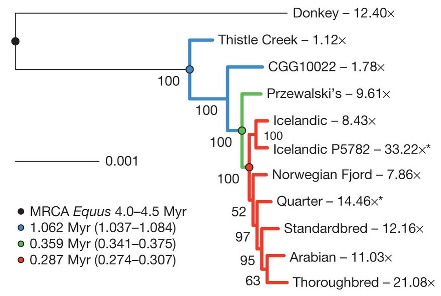
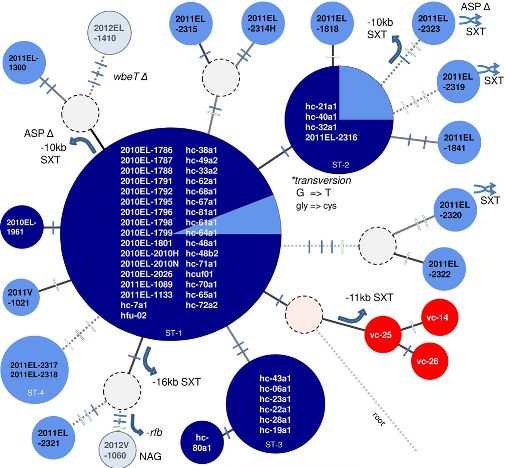
 Dos cepas de virus de influenza aviar H7N9, que ha provocado docenas de muertes humanas en China, han sido secuenciadas de manera independiente por dos equipos de investigadores. Puede leer los reportes completos en Watanabe T, Kiso M, Fukuyama S, Nakajima N, Imai M, Yamada S, et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 10 July 2013; doi:10.1038/nature12392, y Belser JA, Gustin KM, Pearce MB, Maines TR, Zeng H, Pappas C, ET AL. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 10
Dos cepas de virus de influenza aviar H7N9, que ha provocado docenas de muertes humanas en China, han sido secuenciadas de manera independiente por dos equipos de investigadores. Puede leer los reportes completos en Watanabe T, Kiso M, Fukuyama S, Nakajima N, Imai M, Yamada S, et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 10 July 2013; doi:10.1038/nature12392, y Belser JA, Gustin KM, Pearce MB, Maines TR, Zeng H, Pappas C, ET AL. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 10
July 2013; doi:10.1038/nature12391.
Filed under Diagnóstico, Filogenética, Otros Proyectos Genoma, Patógenos by on . Comment. ![]()