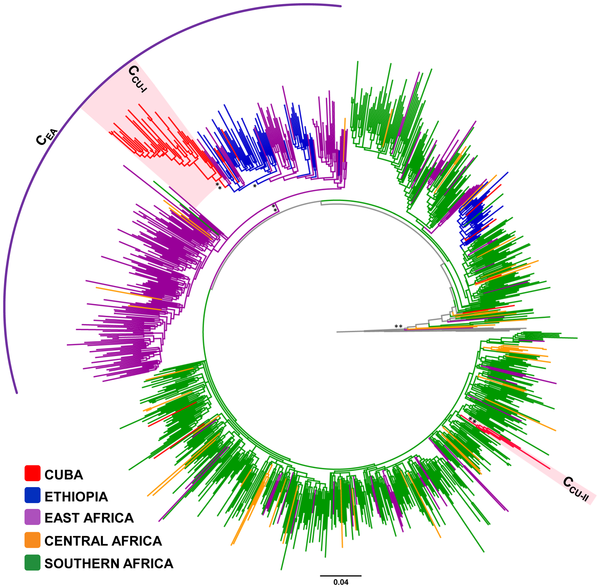
 El análisis filogenético de las cepas de VIH-1 circulantes en Cuba condujo a que la causa de la epidemia en la isla fue la introducción de unas pocas cepas que circularon ampliamente. Los detalles aparecen en Delatorre E, Bello G. Phylodynamics of the HIV-1 Epidemic in Cuba. PLoS ONE 2013;8(9):e72448.
El análisis filogenético de las cepas de VIH-1 circulantes en Cuba condujo a que la causa de la epidemia en la isla fue la introducción de unas pocas cepas que circularon ampliamente. Los detalles aparecen en Delatorre E, Bello G. Phylodynamics of the HIV-1 Epidemic in Cuba. PLoS ONE 2013;8(9):e72448.
Filed under Filogenética, Patógenos by on . Comment. ![]()