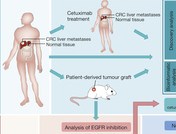
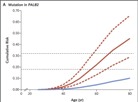
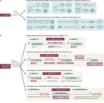
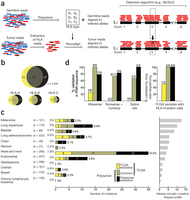
 Cerca de 300 mutaciones no silentes en puntos específicos de los genes de las moléculas HLA-A, -B y -C han sido reveladas por una metodología computacional presentada en Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nature Biotechnology 15 September 2015; doi:10.1038/nbt.3344.
Cerca de 300 mutaciones no silentes en puntos específicos de los genes de las moléculas HLA-A, -B y -C han sido reveladas por una metodología computacional presentada en Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nature Biotechnology 15 September 2015; doi:10.1038/nbt.3344.
Filed under Algoritmos, Bioinformática, Cáncer, Diagnóstico, Inmunoinformática by on . Comment. ![]()